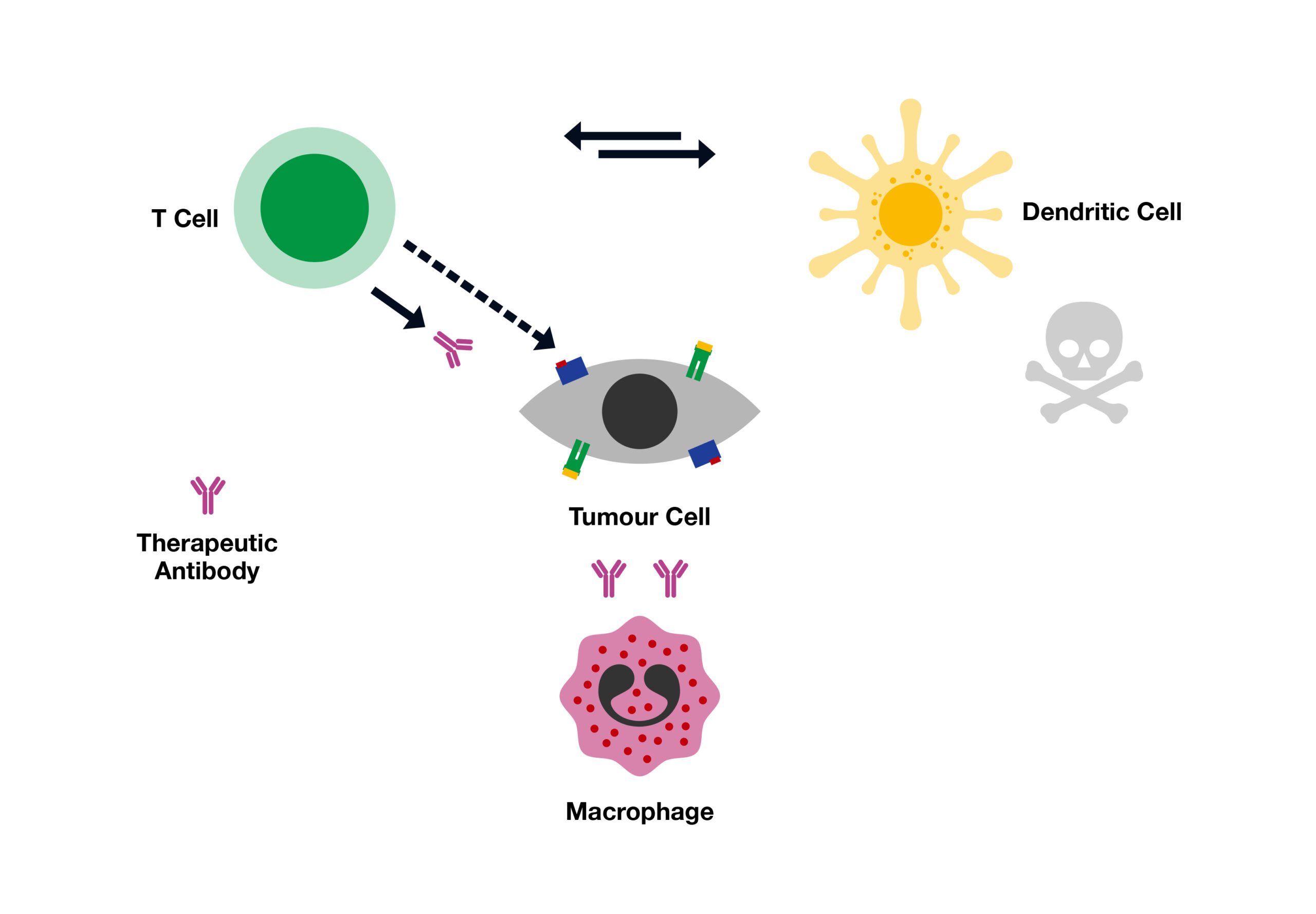
Our immune system is an extraordinary sentinel that protects our body from harmful pathogens such us bacteria and viruses (the innate immunity), produce antibodies against foreign substances and destroy abnormal cells that may evolve into cancer cells (the adaptive immunity).
A balanced immune system can perfectly target and remove cancer cells before its evolution to a detectable tumour. However, an imbalanced immune system response could contribute to tumorigenesis or induce inflammation stimulating cancer cell proliferation and metastasis, leaving these cells growing effectively avoiding our immune system. It was in 2010 that the FDA approved the first immune therapy against prostate cancer, launching de facto the modern immunotherapy.
Cancer immunotherapy or immuno-oncology, by definition, represents a specific targeted therapy by using our immune system to fight and destroy cancer cells. As this does not involve surgery or use of radiation or chemotherapy, it is relatively safe to the healthy cells of the body. Importantly, it could be applicable at all stages of the disease, with higher efficiency.
The whole objective of Immunotherapy is to enable our immune system understand differences between healthy and cancer cells and then specifically target the cancer cells. We now have the tools to produce specific substances that stimulate our immune system to recognise and fight specific cancer cells. Monoclonal antibodies, checkpoint inhibitors and cytokines represent a class of immuno-oncology therapies that trigger an immune response to destroy cancer cells.
In conclusion, immunotherapy (or immuno-oncology) has opened a new era in healthcare and indeed we can soon expect some ground-breaking discoveries that will completely erase our current notion of cancer as a life-threatening disease. Having said that, modelling the immune system in vitro to stimulate an effective response to cancer sets a particularly difficult challenge. Potential roles of multiple immune cells, the heterogeneity of tumours and the molecular mechanisms involved mean that multiple advanced assay models are required before moving the most promising immunotherapeutic approaches into clinical trials.
For a more in depth look at our immuno-oncology expertise, click the button below to gain access to our immuno-oncology hub, with a detailed look at our data sets and offering.
Request a consultation with Cellomatics Biosciences today
Our experienced team of in vitro laboratory scientists will work with you to understand your project and provide a bespoke project plan with a professional, flexible service and a fast turnaround time.
To request a consultation where we can discuss your exact requirements, please contact Cellomatics Biosciences.
We use cookies on our website to give you the most relevant experience by remembering your preferences and repeat visits. By clicking “Accept”, you consent to the use of ALL the cookies. However you may visit Cookie Settings to provide a controlled consent.
This website uses cookies to improve your experience while you navigate through the website. Out of these cookies, the cookies that are categorized as necessary are stored on your browser as they are essential for the working of basic functionalities of the website. We also use third-party cookies that help us analyze and understand how you use this website. These cookies will be stored in your browser only with your consent. You also have the option to opt-out of these cookies. But opting out of some of these cookies may have an effect on your browsing experience.
Necessary cookies are absolutely essential for the website to function properly. These cookies ensure basic functionalities and security features of the website, anonymously.
Cookie
Duration
Description
cookielawinfo-checkbox-analytics
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Analytics".
cookielawinfo-checkbox-functional
11 months
The cookie is set by GDPR cookie consent to record the user consent for the cookies in the category "Functional".
cookielawinfo-checkbox-necessary
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookies is used to store the user consent for the cookies in the category "Necessary".
cookielawinfo-checkbox-others
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Other.
cookielawinfo-checkbox-performance
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Performance".
viewed_cookie_policy
11 months
The cookie is set by the GDPR Cookie Consent plugin and is used to store whether or not user has consented to the use of cookies. It does not store any personal data.
Functional cookies help to perform certain functionalities like sharing the content of the website on social media platforms, collect feedbacks, and other third-party features.
Performance cookies are used to understand and analyze the key performance indexes of the website which helps in delivering a better user experience for the visitors.
Analytical cookies are used to understand how visitors interact with the website. These cookies help provide information on metrics the number of visitors, bounce rate, traffic source, etc.
Advertisement cookies are used to provide visitors with relevant ads and marketing campaigns. These cookies track visitors across websites and collect information to provide customized ads.